
Development of Manufacturing Capability for Rare Sugar Nucleotides
This project will aim to develop both synthetic biology and hardware tools to better understand and utilise the pectic polysaccharides.
The Idea
Plant cell walls are an intricate assembly of polysaccharides and phenolic compounds. There is a significant interest in the use of plant cell walls as a source of energy and to extract compounds which may have industrial application. One of the biggest hurdles in the development of cell wall derived products is our poor understanding of cell wall biosynthesis. Synthesis of polysaccharides occurs mainly through the activity of Glycosyltransferase (GT) enzymes that transfer an activated nucleotide-sugar onto a specific growing polysaccharide acceptor. The ability to manufacture sugar nucleotides is therefore of profound importance to the development of novel technologies for study and engineering of polysaccharide biosynthesis. While many sugar nucleotides are commercially available, some of the key sugar nucleotides employed in the synthesis of pectin are not available. Better understanding of cell wall biosynthesis, enabled through experiments using manufactured sugar nucleotides, may enable us to engineer the polysaccharides to have desired properties. This project will focus on pectin, which is a heterogenous polysaccharide found in plant primary cell walls. Pectin is abundant in food waste and as a highly negatively charged polymer it might find industrial application. We will aim to develop a synthetic biology toolbox to synthetise nucleotide sugars, which is required for in vitro analysis of pectin biosynthesis enzymes and is currently not available commercially.
We propose to develop the technology to manufacture a range of novel sugar nucleotides that are presently not available commercially. This will include sugar nucleotides that are required to study pectin biosynthesis enzymes. Sugar nucleotide biosynthesis occurs in the Golgi apparatus and is performed by a range of different enzymes. As a part of our project we propose to synthetise GoldenGate compatible modules encoding sugar nucleotide biosynthetic enzymes and express those in E. coli. Expressed enzymes will be used to convert commercially available sugar nucleotides into sugar nucleotides that are not available commercially, or that are prohibitively expensive. Sugar nucleotides will then be purified through a combination of enzyme treatments and chromatographic separations.
By doing so we will enable further study into the structure and function of glycosyl transferases and the manufacture of novel pectic saccharides.
The Team



Dr Tom Simmons,
Founder and CEO of Cambridge Glycoscience, Cambridge
Mr Jan Lyczakowski,
Graduate student, Department of Plant Sciences, University of Cambridge
Dr Henry Temple,
Postdoctoral researcher, Department of Plant Sciences, University of Cambridge
Project Outputs

Project Report
Summary of the project's achievements and future plans

Project Proposal
Original proposal and application

Project Resources
Report: Toolbox cloning and UDP-Rhamnose biosynthesis
Summary
Plant cell wall polysaccharides form the largest biorepository of carbon on the planet and are made primarily from cellulose, pectin, hemicelluloses and lignin. Hemicelluloses and pectin are important for the maintenance of cell wall properties and are synthesized in the Golgi apparatus by glycosyltransferases, using sugar nucleotides as a substrate. Research into pectin and hemicellulose structure and biosynthesis for synthetic biological engineering is hindered by poor commercial availability of sugar nucleotides. This project aimed to generate a synthetic biology sugar nucleotide interconversion toolbox. By expressing and purifying active domains of interconverting enzymes we are now in good position to synthesize UDP-Rhamnose (UDP-Rha) from easily available UDP-Glucose (UDP-Glc). In addition to successfully generating the tools needed for the synthesis of a critical sugar nucleotide required for pectin biosynthesis, we have also cloned genes encoding enzymes required for UDP-Galacturonic acid (UDP-GalA) and UDP-Xylose (UDP-Xyl) formation. Together with a high efficiency bacterial expression vector, pHAT, the GoldenGate compatible DNA sequences encoding sugar nucleotide interconverting enzymes form an IP-free toolbox which is now available from the Dupree laboratory. Additionally, by collaborating with an industrial partner, Cambridge Glycoscience, we were able to bring into the OpenPlant community invaluable expertise on sugar nucleotide manufacturing which can benefit other researchers in the glycobiology work package.
Report and outcomes:
Biosynthesis of sugar nucleotides is a multi-step process which in vivo occurs in both Golgi apparatus and in the cytosol. In our project, we have focused on cytosolic UDP-Rhamnose synthase which converts UDP-Glucose to UDP-Rhamnose, and the Golgi localised steps of converting UDP-Glucuronic Acid into UDP-Galacturonic acid and UDP-Xylose. The biosynthesis of these molecules is synthesised by UDP-D-Glucuronate 4 epimerase (GAE), UDP-Xylose synthase (UXS) (see Figure 1 presented after Temple et al., 2016)
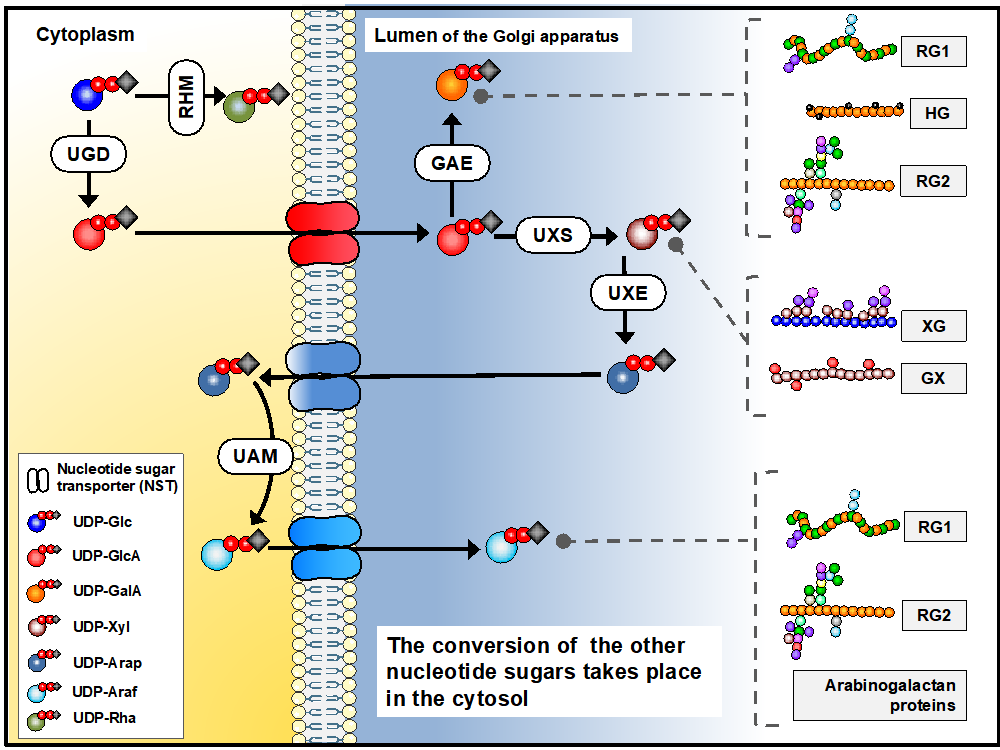
Figure 1. Biosynthesis of sugar nucleotides in plant cells. Sugar nucleotides are required for pectin (RG1, RG2, HG), xyloglucan (XG) and xylan synthesis.
Figure presented after Temple et al., Glycobiology, 26 (9), 913–925.
Toolbox cloning
DNA sequences encoding GAE, UXS and two domains of RHM (domain D and domain ER) were synthesized as GoldenGate compatible linear fragments. DNA synthesis was performed by Integrated DNA Technologies (IDT). Linear fragments were PCR amplified using Q5 DNA polymerase and blunt end cloned into AfeI digested pHAT vector. PCR was used to screen for correct orientation of inserts and DNA sequencing was used to confirm correct insert identity. We were able to successfully clone all genes into the pHAT vector. The sequences encoding the pHAT expression vector, GAE, UXS, RHM-D and RHM-ER were deposited in the OpenPlant Benchling repository in a dedicated sugar nucleotide interconversion toolbox folder. Fasta sequences for all sugar nucleotide interconversion enzymes were also included as Attachment 1 to this report.
Expression of UDP-Rhamnose biosynthesis genes
UDP-Rhamnose synthesis occurs by the activities of two domains of RHM proteins. The first reaction of the interconversion is UDP-Glucose 4,6 dehydratase (N-terminal domain of the protein, RHM-D) and then followed by UDP-4-keto-6-deoxyglucose 3,5-epimerase and UDP-4-keto-rhamnose-4-keto-reductase (C-terminal domain of the protein, RHM-ER). Since the product UDP-Rhamnose is an inhibitor for the UDP-Glucose 4,6 dehydratase activity, we have decided to split the two domains of RHM protein to carry stepwise reactions (D and ER domains).
We have transformed pHAT-RHM-D and pHAT-RHM-ER into Rosetta 2 E. coli cells. We screened expression from 8 individual clones using small scale (5 mL) fermentation with overnight induction using IPTG. The highest expressing clone for each construct was picked and grown in larger scale culture (250 mL). For larger scale protein production, we have performed induction of cell culture in exponential growth phase at 30°C with 0.5mM IPTG for 3h. Following protein expression, we have harvested the cells and lysed them using sonication. Cell lysates were used to perform protein purification using Ni-NTA agarose columns, and we successfully purified two domains of RHM protein (Figure 2).

Figure 2. Expression and purification of domains RHM-D (A) and RHM-ED (B). Good degree of expression and purification was obtained for both proteins.
UDP-Rhamnose synthesis using recombinant enzymes
With purified proteins, we are now able to synthesise UDP-Rha from UDP-Glc. We have performed biochemical assays on the activities of these purified domains. First, we incubated recombinant RHM-D with UDP-Glc overnight, and then incubated the reaction products with recombinant domain RHM-ER. The products were then acid-hydrolysed to cleave the sugar moiety from the UDP component and reaction products were analysed by HPLC. HPLC showed that in the absence of both enzymes, or with enzyme ER alone, only glucose was present, consistent with a lack of conversion activity. In the presence of enzyme D alone only glucose was present, but at a reduced amount, consistent with some conversion of the UDP-Glc substrate into a compound that was either unstable in acid or migrated at an unknown place on the chromatogram. Finally, in the presence of both D and ER, glucose was present at a reduced amount, and an additional peak which migrated with Rhamanose appeared, consistent with some conversion of the UDP-Glc substrate into Rhamnose. Conversion was unfortunately quite low, and we are working to improve this through optimised reaction conditions.
Follow on Plans
We have progress in getting interconverting enzymes for obtaining UDP-Rhamnose from UDP-Glucose, our following step is to continue testing more conditions to obtain optimal sugar nucleotide interconversion.
We have also attempted to express and purify UXS and GAE. However, although we used versions of these proteins without their transmembrane domain, we had very low soluble protein yield and were unable to perform protein purification. We plan to trial more expression conditions, but if this result in unsuccessful efforts, we would like to test other Arabidopsis enzymes encoding the same activities. This might require some more DNA synthesis. If we are able to express UXS and GAE we would like to attempt more sugar nucleotide conversion reactions. This might require additional input from Cambridge Glycoscience. However, we believe that the remaining £1500 should be sufficient to cover the costs of any additional DNA synthesis and conversion experiments. Any new sequences generated will be submitted to the OpenPlant Benchling folder and will be made available to the community on an IP-free basis.