
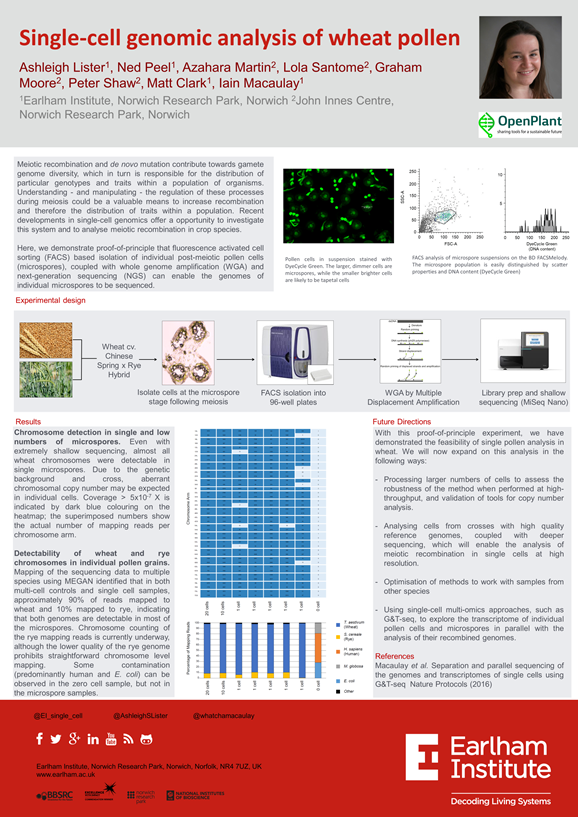
Single cell pollen meiosis screening in wheat
This project will look at wheat meiosis at the single cell level, which has not been done before. The project aims to develop a quick and cheap screening assay to identify meiocytes which we want to look at in more detail (by deeper DNA sequencing) and to discover the level of recombination during wheat meiosis and locate any hot spots.
The Idea
Recombination is a process which takes place during meiosis, a system necessary during gametogenesis in all sexual organisms. The two sets of replicated parental chromosomes undergo crossover formation followed by two rounds of segregation, to produce haploid gametes ready for fertilisation. Variation in the offspring population, caused by meiotic recombination as well as random segregation, is highly important as it creates a fitness in the event of a change in environment or pathogen attack, as well as creating genetic diversity required in agricultural plant breeding.
We would like to screen wheat meiocytes (from a given parent plant) at the single cell level because this will show the extent of meiotic recombination, and any recombination hotspots, without the need to grow hundreds, or thousands of plants. Our method would be a quick, cheap and high-throughput method of scoring new hybrids, and could be used to screen for treatments to affect meiosis. The method would identify which hybrids, or treatments have elevated recombination events, which would be a powerful tool for breeding or programmes and for future research.
The Team





Ms Ashleigh Lister,
Research Assistant, Technical Development, Earlham Institute, Norwich
Dr Iain Macaulay,
Research Group Leader, Technical Development, Earlham Institute, Norwich
Dr Matt Clark,
Head of Technology Development, Earlham Institute, Norwich
Prof Graham Moore,
Research Group Leader, Crop Genetics, John Innes Centre, Norwich
Prof Peter Shaw,
Research Group Leader, Cell and Developmental Biology, John Innes Centre, Norwich
Project Outputs

Project Report
SUMMARY OF THE PROJECT'S ACHIEVEMENTS AND FUTURE PLANS

Project Proposal
Original proposal and application

Project Resources
Find out MORE ABOUT THE PROJECT IN THIS poster and these slides.
Six month progress report
We have added a few helpers to the team to help with bioinformatic analysis of the data generated: Ned Peel (Earlham Institute) and Graham Etherington (Earlham Institute).
Project summary
We aimed to create a high throughput single-cell method to sequence wheat pollen cells at a post-meiotic stage, to check for recombination between hybrid chromosomes. From the same cells the RNA can be studied to study the transcripts being expressed at these stages. We believe that this would be a very beneficial tool to study meiotic systems but also as a genotyping tool for breeding.
Report and outcomes

Wheat grown in a controlled environment room enabling monitoring of temperature and pathogens, were grown until the stage of collection following tetrad stage, when still uninucleate. Around 50 anthers at the same pollen stage were collected into PBS and crushed using a metal rod, releasing the pollen from the internal pollen tube structure and surrounding tapetal cells. The solution was passed through a 100um filter before staining using Dye Cycle Green and loading onto the FACSMelody instrument at the lowest possible flow rate.
FACS plots; we selected our pollen cells if they were fairly big as opposed to the background debris and smaller contaminating cells within the anther preparation, and how well they stained with our Dye Cycle Green. This can be done on the FACSMelody using plots of Side Scatter vs Forward scatter, and side scatter vs green fluorescence. Each population can be sorted and checked under a microspore which verifies the correct cell type and how well they have coped having been sent through the sorter (in comparison to a pre-sort check).


We have sequenced DNA and RNA from the parental lines to provide us with control data ready to look at hybrid material. We have the ability to do this at the Earlham Institute using the Beckman Coulter robotic automation instrumentation and G&TSeq (Macaulay et al, 2016). The purpose of sequencing the parental lines only was two-fold; the hybrid material was not ready yet and, by having no hybridisations there should be no recombination visible in the data therefore any low read counts for certain areas would be as a result of technical drop-out. From this data we can also determine if we have got the depth of genome and transcriptome coverage correct, or if we need more reads per run.
The following graphs are as a result of the transcriptome analysis. Most are quality checks to determine how many reads per sample we achieved.

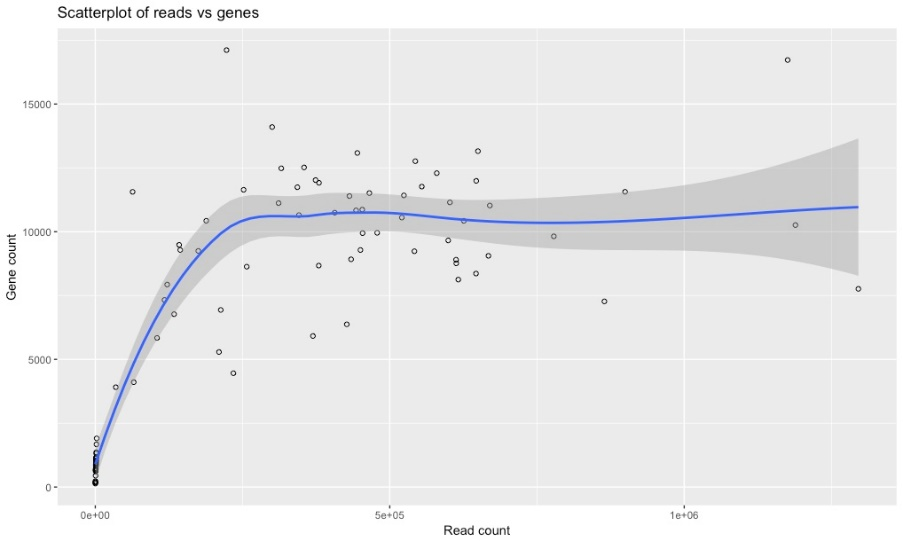

Plate plan showing the number of cells deposited per well.


{kind=link}
The following heatmap plots the number of reads mapped to each chromosome arm in the wheat genome, per single cell sample. This gives us a good visualisation as to if there are any areas of the genome that are underrepresented for reasons such as GC-rich spots, this will be the kind of plot we will look at when we sequence the hybrid pollen to spot any gaps in the genome indicating recombination between the two parental chromosome copies.
In the last few days, the hybrid pollen has been at the correct stage ready for collection. Therefore, we have processed this through to FACS sorting. These cells will be processed as the parental samples above were, by splitting the DNA from the RNA, and constructing libraries from them before sequencing them at fairly low coverage to keep the costs down.
Summary
Although we started out with the intention of sequencing of hybrid pollen, we ran into set-backs after analysing some preliminary data. The hybrid pollen that would have been immediately available to us was the result of a cross, with one of the parents (rye) which had an incomplete genome assembly, therefore, the resolution of the mapping would not be good enough to spot recombination from our DNA libraries. There was a delay as a result of growing a new hybrid, Chinese Spring and Cadenza which had far improved genome assemblies, and waiting for the pollen to be ready.
We have sorted, sequenced and analysed the parental lines of our newly chosen parental lines. Therefore, we feel like at this stage in the project, we have got the technique we proposed to OpenPlant working from pollen staging, sample preparations, FACS sorting, cell lysis, DNA and RNA amplification from single cells, low-cost, low-volume library construction and sequencing, and we have the analysis pipelines in place ready for the hybrid material once it is ready.
Outreach presentations
Presented poster using preliminary MiSeq data at Genome10K and Genome Science conference http://www.earlham.ac.uk/genome-10k-and-genome-science-conference
Presented poster at SAB.
Iain will present project at AGBT conference in Feb 2018, http://www.agbt.org/gm-agenda/
Led to other similar projects, single cell Zebrafish sperm and mouse sperm sorting looking also at meiosis and recombination
Led to a meiosis conference ‘Meiosis and Beyond’ http://www.earlham.ac.uk/meiosis-and-beyond, to be held 5th March 2018
Once the first hybrids have been analysed, we hope to publish both the technique and the biology.
Follow on plans
We still need to sequence the hybrid pollen although this will be completed after I return from maternity leave. We have the pollen sorted into plates ready to go.
We would like to extend the study to look at pollen during different stages of gametogenesis, especially to look at differences within the transcriptome.
The same method can also be applied to different wheat cultivars, hybrids and treatment types.
We would like to use the same method to work with gametes from different species such as Zebrafish and mouse.
References
Iain C Macaulay, Mabel J Teng, Wilfried Haerty, Parveen Kumar, Chris P Ponting, & Thierry Voet, (2016) Separation and parallel sequencing of the genomes and transcriptomes of single cells using G&T-seq, Nature protocols, 11(11) p2081- 2103 doi:10.1038/nprot.2016.138